





DDX1甲基化介導的MATR3 剪接通過啟動染色質重編程來調節椎間盤退化
下腰痛(LBP)主要由椎間盤退變(IVDD)驅動,已成為公共衛生領域的核心挑戰。RNA結合蛋白DDX1在RNA代謝中起關鍵作用,但其在IVDD中的功能尚不明確。本研究發現DDX1是甲基轉移酶EZH2的底物,EZH2對DDX1第234位賴氨酸(K234)的甲基化修飾可促進體外和體內IVDD進程。抑制EZH2能恢復髓核(NP)細胞的基質穩態并延緩IVDD發展。DDX1 K234位點的甲基化會破壞其與剪接因子及RNA靶標的相互作用,導致MATR3基因外顯子14跳躍。由此產生的截短型MATR3會破壞核結構、增加染色質可及性,進而激活Wnt等信號通路,引發NP細胞衰老和凋亡。值得注意的是,通過陽離子脂質納米顆粒遞送過表達MATR3-L的mRNA可顯著減輕NP細胞退變并有效緩解IVDD,這為IVDD的發病機制研究和潛在治療策略提供了重要依據。該研究于2025年7月發表在《Nature Communications》,IF:15.7。
技術路線:
主要研究結果:
1.椎間盤退變過程中髓核細胞DDX1的K234甲基化水平升高
為探究賴氨酸甲基化調控的關鍵非組蛋白[17],本研究獲取腰椎骨折、特發性脊柱側凸或IVDD患者的非退變與退變髓核組織進行細胞培養,裂解后采用泛賴氨酸甲基化抗體孵育,并通過LC-MS/MS進行組分分析(圖1A)。基于MRI影像的Pfirrmann分級評估顯示:隨著IVDD進展,T2加權像的高信號白色區域逐漸減少并呈現異質性,最終被高信號黑色區域取代,提示髓核組織退變[18]。培養基中水分含量下降而纖維化程度增加,番紅O-固綠染色進一步證實退變髓核組織的表型變化——基質降解、鈣化、軟骨樣增生及細胞簇狀聚集現象在嚴重退變組(IV級)尤為顯著[19](附圖1A)。免疫組化結果顯示退變髓核組織中Ⅱ型膠原陽性(合成代謝)細胞減少,而MMP3表達(分解代謝)細胞相應增加(附圖1B、E)。值得注意的是,質譜分析在退變髓核組織中特異性鑒定出DDX1衍生肽段,其SPRY結構域(231-240aa)內進化保守的賴氨酸殘基(K234)存在甲基化修飾(附圖1C)。鑒于SPRY結構域的功能重要性,該位點甲基化可能具有生物學意義。LC-MS證實K234為單甲基化修飾(圖1B),且該位點在多個物種DDX1蛋白中高度保守(附圖1D),暗示其潛在功能價值。在髓核細胞和HEK-293T工具細胞中均檢測到K234甲基化(圖1C、D,附圖1F)。由于DDX1作為RNA結合蛋白,其功能可能受賴氨酸甲基化調控,進而影響細胞內信號通路和基因表達網絡。
為研究氧化應激損傷是否導致DDX1 K234甲基化差異表達[20],我們采用叔丁基過氧化物(TBHP)處理髓核細胞構建體外退變模型。TBHP處理后,Ⅱ型膠原和聚集蛋白聚糖表達下降,而MMP3和ADAMTS5呈現相反趨勢(附圖1H、I)。免疫熒光顯示TBHP處理的髓核細胞中Ⅱ型膠原陽性信號減少、MMP3增加(圖1E,附圖1G),證實該模型能模擬IVDD的退變特征[21,22]。值得注意的是,TBHP處理的髓核細胞和HEK-293T細胞中DDX1 K234甲基化水平顯著升高(圖1F,附圖1J)。為進一步驗證,我們將SPRY結構域第234位賴氨酸替換為精氨酸構建DDX1突變體[23,24]——精氨酸(R)在保留正電荷的同時可阻斷甲基化,從而模擬非甲基化蛋白狀態。在TBHP處理的HEK-293T細胞中,DDX1 K234R突變體始終未檢測到甲基化(圖1G)。這些結果共同證實SPRY結構域Lys234是DDX1特異的甲基化位點,且與髓核細胞退變密切相關。
鑒于K234甲基化是通過非偏向性質譜分析鑒定的內源性修飾,我們進一步探究其單甲基化如何影響DDX1功能并促進IVDD。為驗證SPRY結構域甲基化的生物學效應,我們在DDX1敲低的髓核細胞中外源性表達野生型DDX1(WT)和甲基化缺陷突變體(K234R)。與對照組(siControl)相比,DDX1敲除可逆轉TBHP誘導的髓核細胞合成代謝下降與分解代謝升高;而重新表達DDX1(而非K234R突變體)則顯著加劇TBHP誘導的細胞退變(附圖1K、L)。為避免脫靶效應,我們通過Western blot驗證了另一種siDDX1在髓核細胞中的敲低效率(附圖2B、C),同時檢測到DDX1 WT和K234R突變體的重新過表達量均達到對照組的約兩倍。
為探究賴氨酸甲基化的體內作用,我們建立了SD大鼠針穿刺誘導的IVDD動物模型[25,26]。通過攜帶DDX1靶向shRNA的慢病毒(shDDX1-LV)在體內敲低DDX1后,每周向尾椎間盤髓核區注射shRNA抗性的DDX1 WT-LV或DDX1 K234R-LV,持續4周[27](圖1H、J)。X射線和CT分析顯示,shDDX1-LV顯著緩解尾椎間盤狹窄和軟骨下骨破壞,而重新表達DDX1-LV(非K234R-LV)會導致椎間盤高度進一步喪失(圖1K、L)。更敏感的MRI檢測顯示,DDX1敲低組尾椎間盤T2加權信號增強[28],表明椎間盤水分得以保留;而重新表達野生型(非精氨酸突變體)則導致水分減少(圖1M)。HE和SO&FG染色證實,慢病毒介導的DDX1缺失可緩解椎間盤退變表型——改善退變椎間盤中紊亂的細胞外基質(ECM)、纖維環(AF)與髓核的模糊界面,并減少纖維環內炎癥細胞浸潤(圖1I、N、O,附圖1M)。免疫組化顯示shDDX1-LV組椎間盤Ⅱ型膠原增加而MMP3減少,提示IVDD進程被抑制(附圖1N、O)。值得注意的是,重新注射DDX1 WT-LV(非K234R-LV)會導致髓核組織萎縮、髓核與纖維環邊界模糊、基質結構紊亂,并加速尾椎間盤進行性退變(圖1I)。此外,野生型DDX1的重新表達(非K234R突變體)還引起Ⅱ型膠原陽性髓核細胞減少和MMP3陽性細胞增加(附圖1N、O)。通過免疫組化驗證另一種shDDX1-LV在椎間盤中的敲低效率以排除脫靶效應(附圖2H、I),同時檢測到DDX1 K234R-LV按預期實現過表達(約為對照組的2倍)。這些結果共同證實,DDX1 K234甲基化可能通過誘導髓核退變在體內外促進IVDD進展。
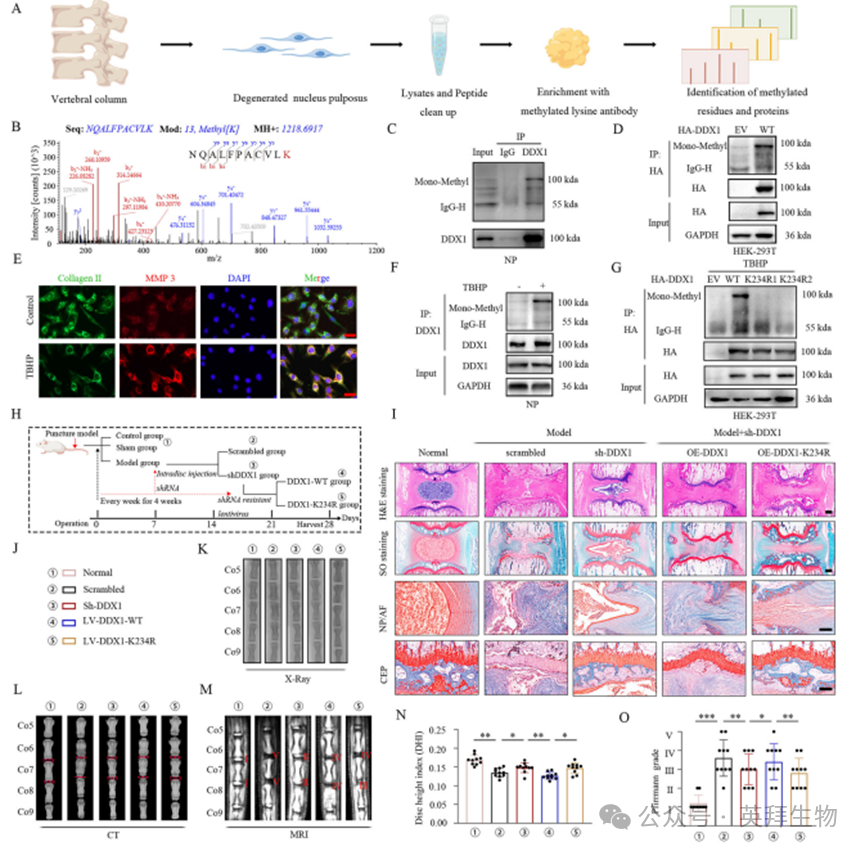
圖1.椎間盤退變過程中髓核細胞DDX1的K234甲基化水平升高
2.DDX1通過EZH2介導的賴氨酸234位點甲基化
為鑒定調控該修飾的生理性甲基轉移酶,我們在HEK-293T細胞中進行免疫共沉淀(Co-IP)實驗篩選DDX1互作蛋白。根據豐度>10且P值<0.05的標準,確定KMT5A、SMYD3、SETD7、EZH2和G9a為候選賴氨酸甲基轉移酶(圖2A、B)。隨后在EZH2質粒轉染的HEK-293T細胞中,抗DDX1免疫沉淀物顯示出顯著的賴氨酸甲基化信號(圖2C)。通過Flag抗體IP結合DDX1免疫印跡(IB)分析發現,EZH2與DDX1的相互作用最強(圖2D)。
EZH2在骨關節炎和類風濕性關節炎中均存在異常激活[29,30],但其在IVDD中的調控作用尚不明確。我們發現,TBHP誘導的髓核細胞體外退變模型中,氧化應激可顯著上調EZH2表達(圖2E、I,附圖2F)。免疫組化顯示退變髓核組織中EZH2陽性細胞呈梯度增加(圖2G、H),提示IVDD進程中髓核細胞的退變狀態與EZH2表達升高相關。免疫熒光證實TBHP處理的退變髓核細胞中,DDX1與EZH2在細胞核內共定位(圖2F,附圖2G)。
進一步通過His標簽DDX1與GST-EZH2的pull-down實驗驗證了兩者的直接物理結合(圖2L,附圖2A)。在髓核細胞和HEK-293T細胞中,內源性與外源性DDX1均能與EZH2強烈互作(圖2J、K、N),表明二者存在直接關聯。這種互作提示甲基化調控的可能性——siEZH2處理的髓核細胞中,泛甲基化抗體檢測顯示DDX1甲基化水平降低(圖2O);反之,EZH2過表達細胞中DDX1賴氨酸甲基化顯著增強(圖2P)。
鑒于DDX1在SPRY結構域K234位點發生單甲基化,我們預測EZH2的甲基化位點即為此位點。通過將賴氨酸234替換為精氨酸構建DDX1突變體,Co-IP結合抗賴氨酸甲基化抗體證實該突變顯著減弱DDX1甲基化(圖2Q)。值得注意的是,K234R和K234M突變均未影響EZH2與DDX1的相互作用(圖2M),這表明EZH2作為DDX1的甲基轉移酶,特異性負責其K234位點的甲基化修飾。
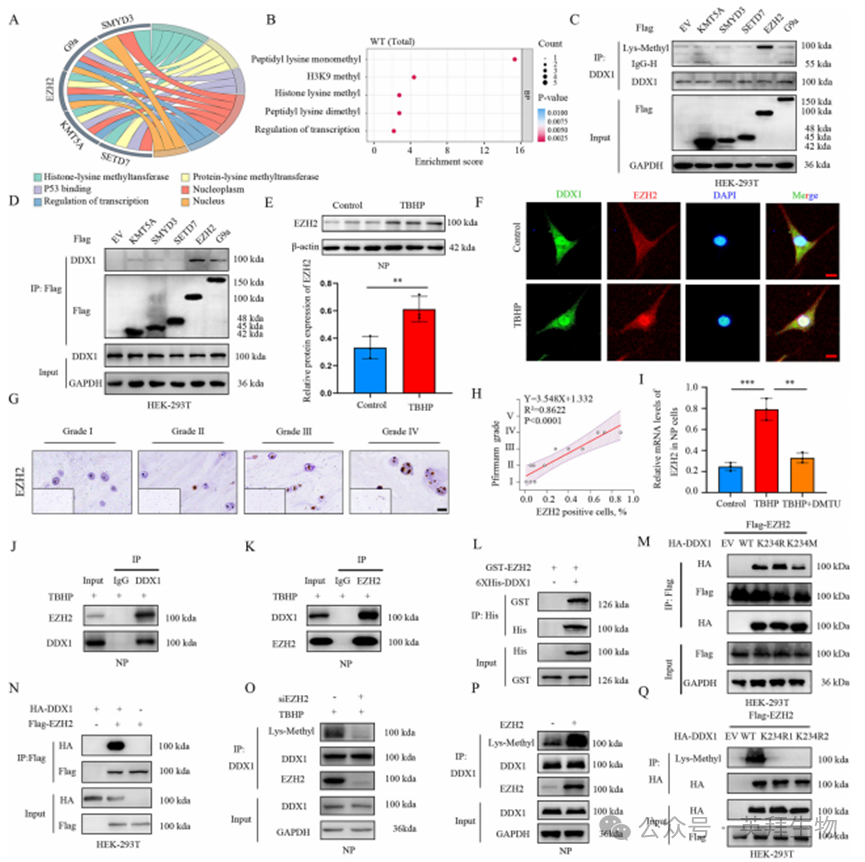
圖2.DDX1通過EZH2介導的賴氨酸234位點甲基化
3.EZH2介導的DDX1 K234甲基化促進椎間盤退變進程
為探究EZH2依賴性DDX1甲基化在IVDD調控中的作用,我們在NP/siDDX1細胞中重新表達DDX1 WT或K234M突變體。值得注意的是,賴氨酸(K)替換為甲硫氨酸(M)可模擬蛋白質的甲基化狀態[13,14,23]。與對照組相比,EZH2敲低能逆轉TBHP誘導的髓核細胞合成代謝下降與分解代謝升高;而重新表達DDX1 K234M突變體(非野生型)則顯著加劇TBHP驅動的髓核細胞退變(圖3A-D)。
為深入解析EZH2依賴性DDX1甲基化在IVDD中的影響,我們采用shEZH2慢病毒敲低SD大鼠體內EZH2后,每周向尾椎間盤髓核區注射DDX1 WT-LV或K234M-LV,持續4周(圖3E、I)。實驗顯示shEZH2-LV顯著緩解尾椎間盤高度丟失和軟骨下骨破壞,而重新表達DDX1 K234M-LV(非野生型)會導致椎間盤高度進一步喪失(圖3F、G、J)。MRI檢測表明EZH2敲低后椎間盤T2加權信號增強(提示水分保留),但K234M突變體重表達(非野生型)則引起水分丟失(圖3H)。HE與SO&FG染色證實,慢病毒介導的EZH2缺失可挽救尾椎間盤退變表型——改善纖維環與髓核間紊亂的ECM結構及模糊界面,并減少纖維環內炎癥細胞浸潤(圖3K-M)。
與體內結果一致,shEZH2-LV處理的椎間盤表現為Ⅱ型膠原陽性細胞增加和MMP3陽性細胞減少,提示IVDD進程被抑制(圖3N、O)。關鍵的是,重新注射攜帶DDX1 K234M突變體的慢病毒(非野生型)會導致髓核組織萎縮、髓核-纖維環邊界模糊、ECM結構破壞,并加速尾椎間盤退變(圖3K)。此外,K234M突變體(非野生型)的重表達會抑制Ⅱ型膠原陽性細胞而促進MMP3陽性細胞(圖3N、O)。這些結果證實,EZH2依賴性DDX1甲基化可通過誘導髓核退變在體內外促進IVDD進展。
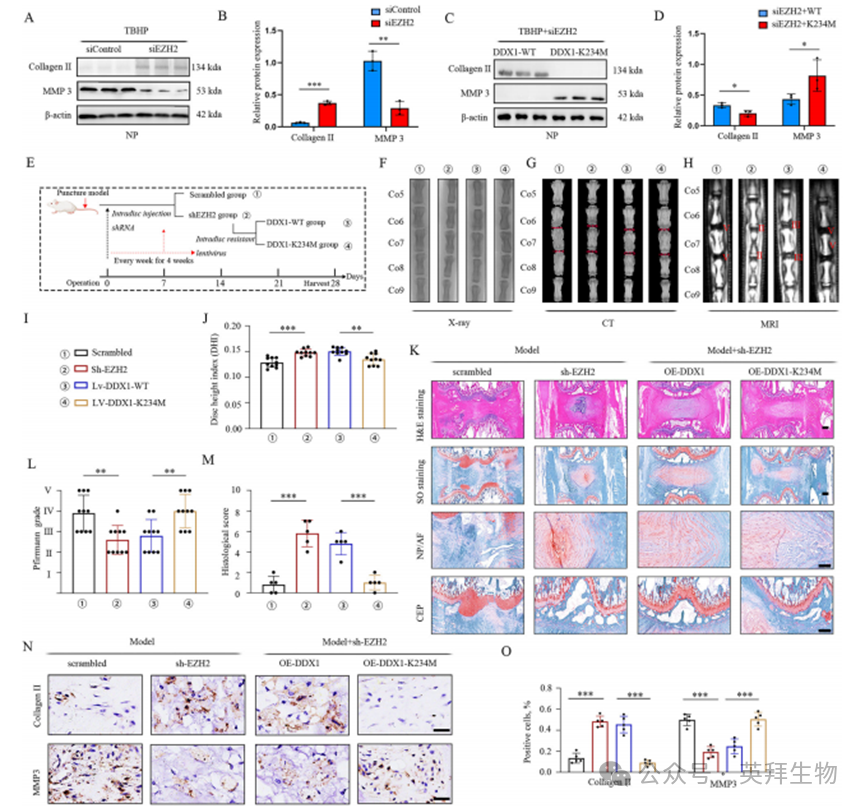
圖3.EZH2介導的DDX1 K234甲基化促進椎間盤退變進程
4.DDX1甲基化促進細胞衰老和凋亡
為了闡明DDX1甲基化的功能意義,我們進行了mRNA轉錄組測序(bulk RNA-seq)分析,比較了正常培養和TBHP培養的NP細胞,以及重新表達siRNA抗性DDX1 K234M或DDX1 K234R突變的NP/siDDX1和HEK-293T/siDDX1細胞的基因集合(圖4A)(補充數據1)。與對照組相比,在TBHP培養的NP細胞中共鑒定出3205個下調基因和3618個上調基因(補充圖3A)。與DDX1 K234R相比,在DDX1 K234M組的HEK-293T細胞中確定了217個下調基因和172個上調基因(補充圖3B、D)。與DDX1 K234R組相比,在DDX1 K234M組的NP細胞中鑒定出349個下調基因(P < 0.05)和486個上調基因(P < 0.05)(補充圖3C)。綜合分析顯示,正常NP細胞中66個上調基因在過表達DDX1 K234R的NP和HEK-293T細胞中重疊(補充圖3E),TBHP處理的細胞中71個上調基因在過表達DDX1 K234M的NP和HEK-293T細胞中重疊(補充圖3F)。隨后,GSEA揭示了細胞衰老和凋亡通路與TBHP處理的NP細胞退行性表型之間的明確相關性(圖4B)。有趣的是,GSEA表明DDX1 K234M過表達同樣調控了細胞衰老和凋亡信號通路(圖4C,補充圖4A、C)。GO分析顯示,DDX1 K234M介導的基因富集于有絲分裂細胞周期、凋亡調控、G2/M轉換、細胞增殖和分裂(圖4D,補充圖4B、D)。先前的研究已在多種細胞類型(包括NP細胞)中鑒定出數百至數千個衰老和凋亡相關基因。其中,與對照組相比,TBHP刺激的NP細胞中有29個經典的衰老和凋亡相關基因表達升高(圖4E)。值得注意的是,這些基因(AIFM1、TNFRSF10A、RELA、BAX、BCL2L1)的表達水平在NP/siDDX1細胞和HEK-293T/siDDX1細胞中重新表達siRNA抗性DDX1 K234M與DDX1 K234R相比具有相似的趨勢(圖4F,補充圖4E)(補充數據2)。這些結果表明,高水平的DDX1 K234甲基化可能在NP細胞中發揮促衰老和促凋亡作用。
與此一致,我們接下來通過Annexin V-FITC/碘化丙啶-PE染色和流式細胞術(FC)檢測了DDX1 K234甲基化介導的NP細胞和HEK-293T細胞凋亡。結果顯示,與對照組相比,siDDX1在體外顯著抑制了NP細胞凋亡。然而,重新表達siRNA抗性的正常DDX1(而非K234R突變體)明顯加劇了TBHP誘導的NP細胞凋亡(圖4G、J,補充圖4G)。HEK-293T細胞中也觀察到類似現象(圖4H、J)。
進一步通過TUNEL和P53染色發現:DDX1敲低可抑制TBHP刺激的NP細胞衰老與凋亡,而重新表達野生型DDX1(非K234R突變體)會促進這兩個過程(補充圖3H、K)。Western blot分析顯示,DDX1失活降低了CASP3和Bax水平,同時增加Bcl-2表達;而過表達野生型DDX1(非K234R突變體)則產生相反效應(補充圖3G、I)。
此外,siRNA敲低DDX1減少了TBHP刺激NP細胞中SA-β-gal染色陽性率,而過表達野生型DDX1(非K234R突變體)增加了SA-β-gal陽性細胞,表明DDX1甲基化促進溶酶體衰老活性(圖4I,補充圖4F)。蛋白水平分析進一步證實,與突變體相比,轉染野生型DDX1的NP細胞中衰老相關蛋白表達增加(補充圖3J、L)。
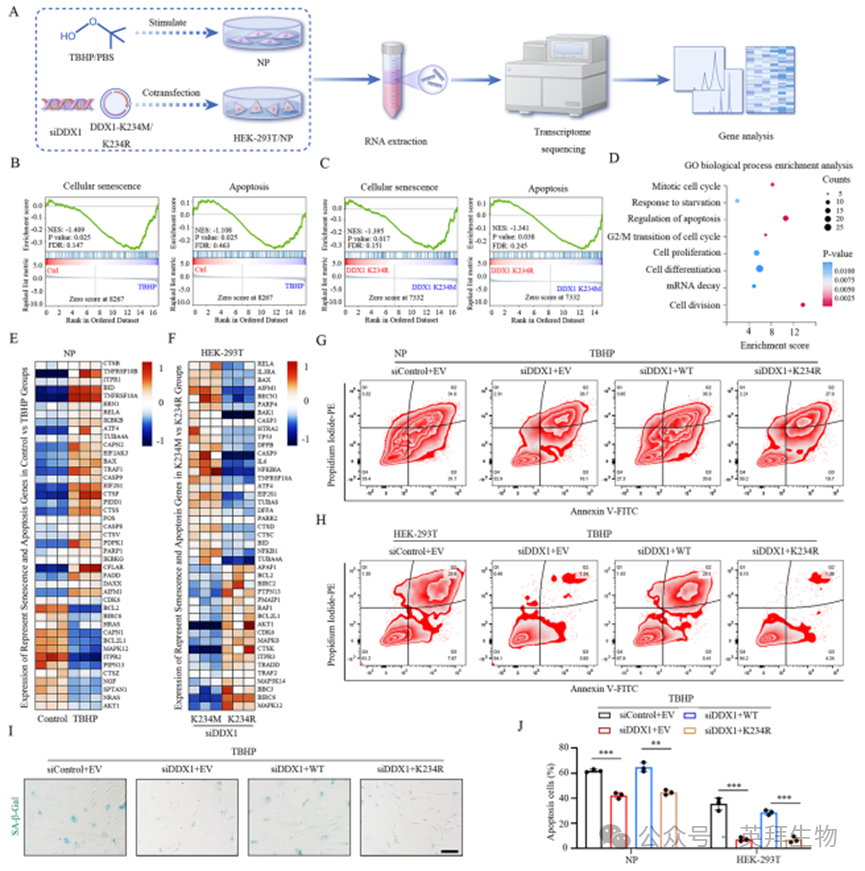
圖4. DDX1甲基化促進細胞衰老和凋亡
5.甲基化導致DDX1在RNA上的結合位點轉錄組減少
為了構建賴氨酸甲基化介導的DDX1-RNA相互作用的基因組圖譜,我們在正常和TBHP處理的髓核細胞中對正常和甲基化DDX1進行了RNA免疫沉淀結合新一代測序(RIP-seq)[33]。對照RIP組共獲得6,724,986條reads,而TBHP-RIP組獲得5,783,158條reads。值得注意的是,Ctrl-RIP組中93.75%的reads和TBHP-RIP組中95.86%的reads被比對到注釋的人類基因組上。由于RNA降解,可能無法在轉錄本上獲得reads的均勻分布,這表明與非結合位點相比,蛋白質在其直接結合位點相對富集,這也使得RIP-seq可以通過peak calling進行分析。peak分布分析顯示DDX1在外顯子、3'UTR和5'UTR區域富集(圖5A、B),最高富集位于外顯子5'UTR末端附近(圖5C、G)。RIP-seq共在對照組和退變組髓核細胞轉錄本中分別鑒定出22,477和10,517個DDX1富集peak(排除了轉錄本自發表達減少引起的誤差)(圖5D)。隨后使用RPKM對基因表達進行定量。聚類分析顯示,與未甲基化DDX1相比,甲基化DDX1結合的注釋人類基因數量減少了約2000個(圖5E)。考慮到非特異性序列結合[34],蛋白質-RNA相互作用位點分析表明,相對于TBHP-RIP,Ctrl-RIP在共享結合區域表現出更強的富集(圖5F、G)。RIP-seq與RNA-seq數據的相關性探索進一步表明,DDX1優先結合在外顯子-內含子連接處附近,特別是在3'和5'剪接位點周圍,其結合強度受賴氨酸甲基化調節(圖5H)。
為闡明DDX1富集峰的功能意義,GO分析顯示甲基化介導的DDX1差異靶基因主要定位于細胞質核周區及核仁區域,這些基因通過結合多種底物在DNA損傷修復、細胞分裂及細胞周期等過程中顯著富集(圖5I、J、M)。為進一步解析DDX1-RNA相互作用,采用HOMER算法鑒定了DDX1結合的RNA基序,發現AGUGGAA七聚體[35]是最顯著富集的元件(圖5K、L、P)。值得注意的是,超過81.5%的這些基序位于外顯子和內含子區域內(圖5N)。在得分最高的四個基序中,基序1和4富集于5'剪接位點的內含子-外顯子連接處附近,而基序2和3則定位于外顯子內部(圖5O)。這些結果共同表明賴氨酸甲基化會減弱DDX1與前mRNA底物的直接結合能力。
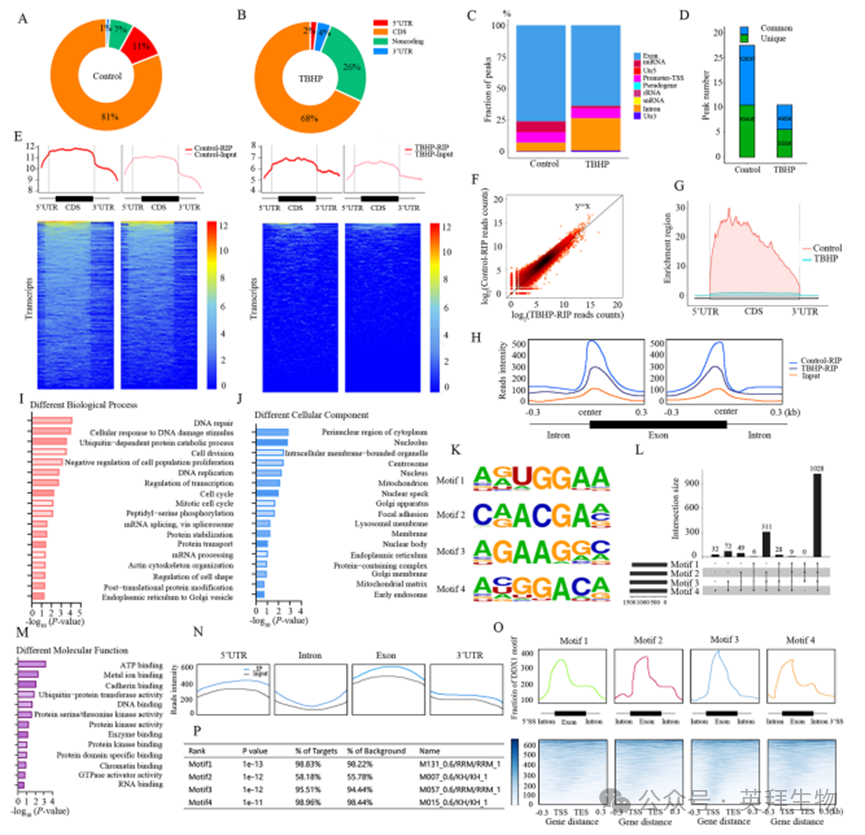
圖5. 單細胞轉錄組測序揭示ASH1L調控轉移性骨微環境中巨噬細胞可塑性
6.甲基化DDX1減少剪接因子招募并促進MATR3外顯子14跳躍
為探究DDX1調控的可變剪接(AS),我們對正常和TBHP處理的NP細胞、以及重新表達siRNA抗性DDX1 K234M和K234R突變的HEK-293T/siDDX1與NP/siDDX1細胞進行了轉錄組分析。AS變異事件包括外顯子跳躍(SE)、內含子保留(RI)、5'和3'位點選擇性切換(A5SS和A3SS)以及外顯子互斥(MEX)表達模式(圖6A)。在TBHP處理的NP細胞中,外顯子跳躍事件占主導(72%),而在過表達DDX1 K234M的HEK-293T細胞和NP細胞中分別降至68%和67%(圖6B,補充圖5A)。值得注意的是,兩類基因譜中第二常見的AS事件均為占比相同(17%)的互斥外顯子。總體而言,NP細胞與HEK-293T細胞中各類AS事件比例相似。
通過整合DDX1結合基因與SE相關基因的轉錄組及RIP數據,我們篩選出與椎間盤退變(IVDD)相關的功能靶點。綜合分析顯示,僅六個外顯子跳躍基因在TBHP處理的NP細胞、以及過表達DDX1 K234M的NP和HEK-293T細胞中與DDX1結合存在重疊(圖6C)(補充數據3)。鑒于RNA-seq結合功能實驗已證實DDX1賴氨酸甲基化通過激活衰老和凋亡信號通路導致NP細胞退變,同時基于RIP-seq的GO分析發現DDX1靶基因富集于核周區域的生物學過程(BP),如DNA損傷修復、細胞分裂與周期調控(圖5I,J)。
在DDX1結合且發生可變剪接的轉錄本中,核基質蛋白編碼基因MATR3(Matrin3)引起了我們的關注。Matrin3蛋白在細胞核內具有維持核基質結構、調控基因轉錄、參與mRNA代謝、DNA修復及凋亡調控等關鍵功能,其異常可能加劇多種疾病發生發展。RNA-seq分析發現,TBHP處理和DDX1 K234M過表達(NP與HEK-293T細胞)均促進外顯子14跳躍,產生更短的MATR3亞型(MATR3-S)。RIP-seq數據進一步顯示DDX1結合于MATR3外顯子14鄰近區域,證實MATR3是DDX1的直接靶標(圖6D,E;補充圖5C,D)。
序列分析表明,外顯子14跳躍導致讀碼框移位但未引入提前終止密碼子,提示該亞型通過結構而非表達量變化影響功能(圖6F)。使用亞型特異性引物檢測發現,TBHP處理顯著降低NP細胞中MATR3-L/MATR3-S比值,DDX1 K234M過表達產生相同效應(圖6G,補充圖5B)。半定量RT-PCR證實TBHP處理增加外顯子14跳躍率,生成更多MATR3-S而減少全長亞型,該現象在DDX1 K234M過表達組中同樣存在(圖6H,補充圖5E)。這些結果共同證實DDX1通過直接結合pre-mRNA調控MATR3可變剪接。
為闡明DDX1調控可變剪接(AS)的機制,我們在過表達DDX1 K234R和K234M突變的siDDX1/HEK-293T細胞中,通過DDX1抗體免疫共沉淀結合質譜(MS)進行分析。GO富集分析顯示,DDX1互作蛋白顯著富集于剪接體介導的mRNA剪接及RNA剪接調控相關通路(圖6I)。與甲基化DDX1相比,正常DDX1中多種RNA剪接因子(如HNRNPA2B1、RBM26和SF3B2)豐度更高(圖6J,補充圖5F-H)。已知DDX1互作蛋白HNRNPK2和DHX36在兩種條件下均被富集,驗證了數據可靠性(圖6J)。
RNA解旋酶通過水解ATP獲得能量來解開RNA雙鏈或二級結構。通過檢測反應體系中ATP的消耗速率,可間接評估解旋酶活性或RNA結合蛋白對解鏈過程的調控作用。K234R和K234M突變體對非特異性結合RNA探針的解旋酶活性無影響,但DDX1 K234M與MATR3的相互作用較野生型(WT)和K234R顯著減弱(補充圖5I)。值得注意的是,基于GO和KEGG分析發現,DDX1 K234R鄰近蛋白在mRNA剪接通路中呈現更高富集度(圖6K-M),而rRNA加工相關蛋白在K234R與K234M間無顯著差異,表明DDX1與剪接調控因子的相互作用獨立于其解旋酶活性。這些結果共同證明,甲基化通過阻礙DDX1招募mRNA加工相關蛋白來調控可變剪接過程
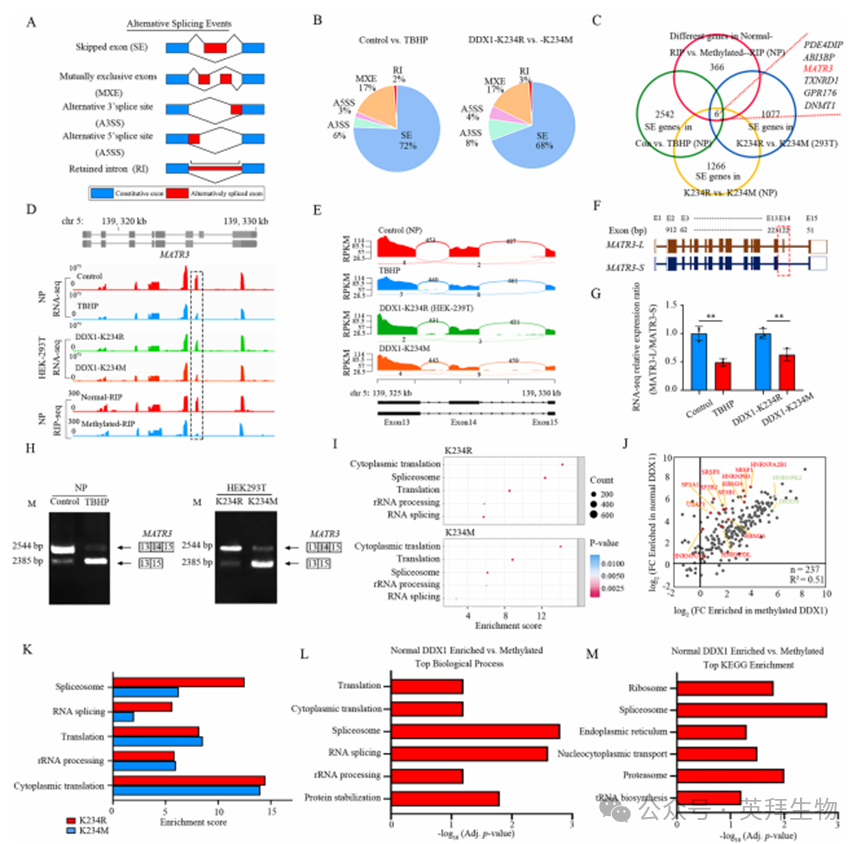
圖6.甲基化DDX1減少剪接因子招募并促進MATR3外顯子14跳躍
7.MATR3-S通過觸發染色質過度開放參與衰老和凋亡區域調控
為評估MATR3外顯子14跳躍的功能意義,我們通過ATAC-seq檢測了過表達MATR3-L(含外顯子14)或MATR3-S(缺失外顯子14)的NP細胞基因組染色質可及性。兩種NP細胞的ATAC-seq數據集均顯示約200 bp的相似片段長度,雙峰分布提示存在不同的染色質結構或特定DNA序列區域(圖7A)。與轉錄研究一致,過表達MATR3-S的NP細胞染色質peak變化數量顯著多于MATR3-L組,且MATR3-L組人類NP細胞中重復元件的ATAC peak信號較弱,表明其染色質結構更為致密(圖7B,補充圖6A)。基于可及染色質peak的主成分分析(PCA)和相關分析明確區分了兩類NP細胞(補充圖6B、C)。染色質peak主要定位于內含子、外顯子、啟動子和遠端區域(通常包含增強子等非編碼調控元件)(圖7C)。值得注意的是,NP細胞中受MATR3亞型變化影響的染色質peak幾乎全部位于遠端區域(補充圖6D),提示非編碼調控元件對MATR3亞型變化具有特殊響應性。研究發現,MATR3亞型對NP細胞染色質可及性動態變化具有相反效應:隨著MATR3-L增加,多數動態染色質peak呈現關閉狀態;而MATR3-S增加則導致這些peak開放(圖7D-F)。這些數據表明,在NP細胞退變過程中,MATR3-L誘導抑制性染色質狀態,而MATR3-S導致染色質結構松弛化。網絡分析揭示Wnt通路作為細胞周期、凋亡和分化等生物過程的核心調控樞紐,與MAPK通路存在復雜交叉對話(圖7G)。β-連環蛋白(β-catenin)激活后可轉位并與轉錄因子互作,促進Cyclin D1等細胞周期基因表達,驅動細胞進入S期。雖然Wnt/β-catenin通常促進細胞存活與增殖,但其過度激活在某些條件下可誘導凋亡。
GO和KEGG分析顯示,過表達MATR3-S的退變NP細胞中,衰老與凋亡相關區域(包括"應激激活的MAPK級聯"、"Ras蛋白信號轉導"、"Wnt信號通路"及"Wnt介導的細胞間信號")的染色質可及性普遍升高(圖7I,補充圖6E)。相反,過表達MATR3-L的正常NP細胞中,抑制細胞周期與生長發育相關區域(如"有絲分裂細胞周期G/M轉換"、"細胞形態發生"和"骨骼系統發育")的染色質可及性顯著降低(圖7J,補充圖6F)。網絡分析揭示Wnt通路作為細胞周期、凋亡和分化等生物過程的核心調控樞紐,與MAPK通路存在復雜交叉對話(圖7G)。β-連環蛋白(β-catenin)激活后可轉位并與轉錄因子互作,促進Cyclin D1等細胞周期基因表達,驅動細胞進入S期。雖然Wnt/β-catenin通常促進細胞存活與增殖,但其過度激活在某些條件下可誘導凋亡。
GO和KEGG分析顯示,過表達MATR3-S的退變NP細胞中,衰老與凋亡相關區域(包括"應激激活的MAPK級聯"、"Ras蛋白信號轉導"、"Wnt信號通路"及"Wnt介導的細胞間信號")的染色質可及性普遍升高(圖7I,補充圖6E)。相反,過表達MATR3-L的正常NP細胞中,抑制細胞周期與生長發育相關區域(如"有絲分裂細胞周期G/M轉換"、"細胞形態發生"和"骨骼系統發育")的染色質可及性顯著降低(圖7J,補充圖6F)。具體而言,過表達MATR3-S的NP細胞呈現以下特征:Wnt通路相關區域(如WNT2/3、CTNNB1、GSK3B)染色質開放度增加(圖7H、K-N;補充圖6J);MAPK通路成員(MAPK1/3)調控區域可及性上升(圖7K);促凋亡基因(RIPK1、CASPASE3)調控元件可及性增強(補充圖6K、M);細胞周期相關基因(如CDK1)染色質可及性下降(補充圖6L)。而過表達MATR3-L的NP細胞在上述調控區域呈現完全相反的染色質可及性變化(補充圖6G-I)。這些結果表明,MATR3不同亞型通過塑造相反的染色質景觀,對衰老和凋亡相關調控區域產生拮抗性調控作用。
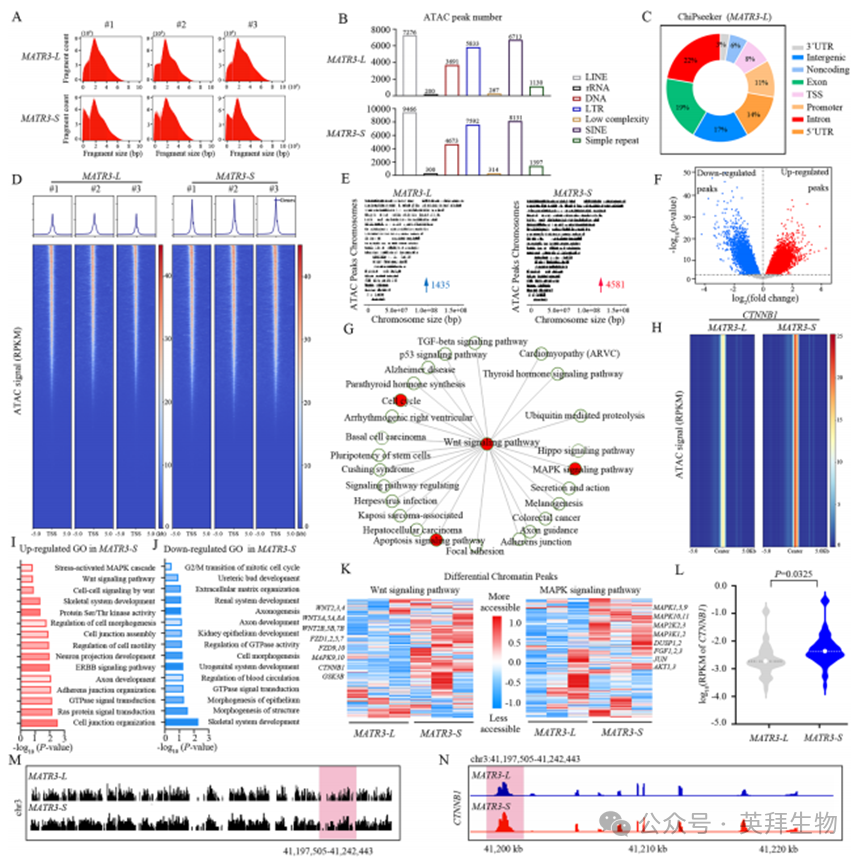
圖7. MATR3-S通過觸發染色質過度開放參與衰老和凋亡區域調控
8.陽離子LNP遞送MATR3-L mRNA有效延緩椎間盤退變進程
鑒于MATR3核苷酸序列較長且可行包裝方案有限,結合脂質納米顆粒(LNP)在相鄰細胞間側向轉運中的優異性能,我們基于微流控技術構建了過表達MATR3-L或MATR3-S的陽離子脂質LNP(補充圖7A)。微流控技術通過精確分配納升級體積、以軸向擴散混合為主及低體積連續操作等特性,可有效控制粒徑并生成多種LNP類型。該體系中:DOTAP通過靜電相互作用直接結合細胞表面,膽固醇增強脂質穩定性,DOPE提高核酸遞送效率,聚乙二醇(PEG)則通過限制血漿蛋白結合與非特異性攝取延長體內循環半衰期。
為構建功能性mRNA轉錄本,我們制備了含MATR3基因的DNA載體。轉錄產物中MATR3-L長度約2544個核苷酸,MATR3-S約2385個核苷酸,與預測長度一致。透射電鏡(TEM)顯示MATR3-L-LNP、MATR3-S-LNP及空載體LNP均呈分散良好的不規則球形或杯狀形態,與其粒徑分布相符(圖8A)。為評估mRNA轉錄本克服核酸酶敏感性、抗原呈遞障礙及載體介導遞送效率等挑戰的能力,瓊脂糖凝膠電泳證實當陽離子脂質體與mRNA重量比(w/w)>8:1時,MATR3-L與MATR3-S可實現完全包裹(圖8B)。納米顆粒追蹤分析(NTA)顯示:空載體LNP平均粒徑80.94±12.28 nm,Zeta電位59.56±5.41 mV;MATR3-L-LNP平均粒徑193.73±10.30 nm,Zeta電位27.56±7.81 mV;MATR3-S-LNP平均粒徑168.19±11.61 nm,Zeta電位35.78±6.11 mV(圖8C、D)。
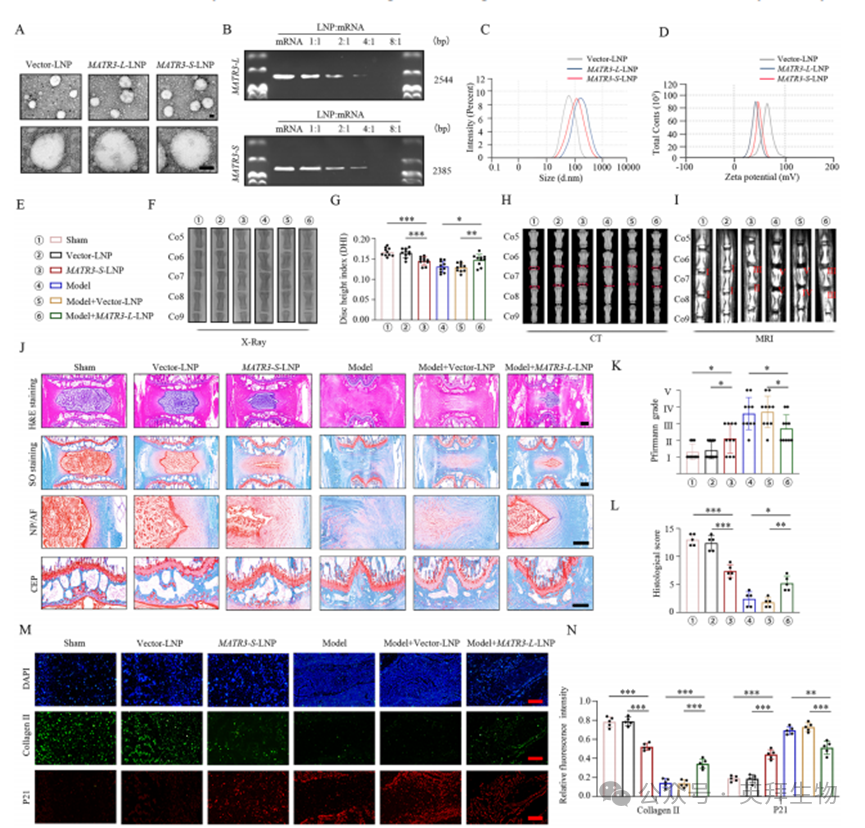
圖8. 陽離子LNP遞送MATR3-L mRNA有效延緩椎間盤退變進程
8.分子機制
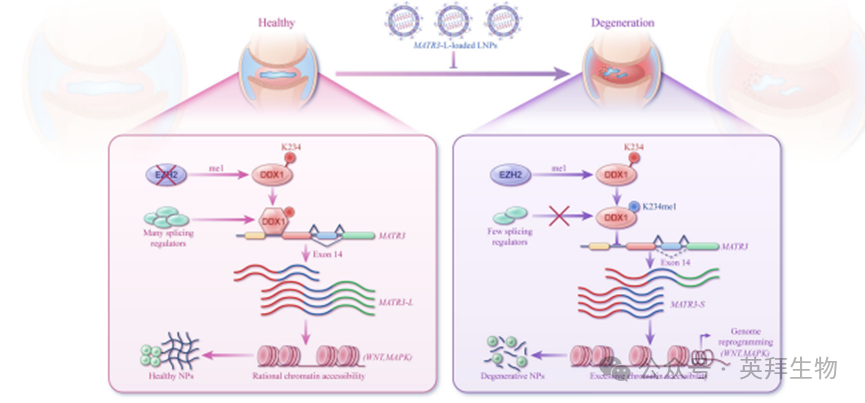
在退變的髓核細胞中,上調的EZH2增強了DDX1第234位賴氨酸的甲基化修飾。甲基化DDX1與MATR3的相互作用減弱,導致剪接位點處剪接因子富集度降低,從而通過外顯子14跳躍促進MATR3-S亞型的生成。MATR3-S通過過度開放染色質可及性,異常激活Wnt信號通路進而促進髓核細胞衰老與凋亡。基于陽離子脂質納米顆粒遞送過表達MATR3-L mRNA的治療策略,可有效延緩椎間盤退變進程。
結論:
本研究發現EZH2介導的RNA結合蛋白DDX1在賴氨酸234位點的甲基化,通過削弱其與剪接因子的相互作用,促進MATR3基因外顯子14跳躍,產生短型MATR3-S,進而引發椎間盤髓核細胞染色質過度開放,激活Wnt等衰老凋亡信號通路,加速椎間盤退變;而利用陽離子脂質納米顆粒遞送MATR3長型mRNA可逆轉這一過程,為椎間盤退變的基因治療提供了新策略。
參考文獻:
Zhu D, Liang H, Tong B, Du Z, Li G, Zhang W, Wu D, Zhou X, Lei J, Zhang X, Ma L, Wang B, Feng X, Wang K, Tan L, Song Y, Yang C. DDX1 methylation mediated MATR3 splicing regulates intervertebral disc degeneration by initiating chromatin reprogramming. Nat Commun. 2025 Jul 4;16(1):6153.